罕见疾病 全球十大罕见疾病,超过一百万人遭受困扰
【www.jxtldy.com--影评书评】
罕见疾病【篇一】罕见疾病
全球十大罕见疾病 超过一百万人遭受困扰 疾病的存在及其分化,可能引导我们穿越整个生命过程。有超过一百万的人们在遭受全球十大罕见疾病的困扰。疾病的存在及其分化,可能引导我们穿越整个生命过程。有超过一百万的人们在遭受全球十大罕见疾病的困扰。

吸血鬼症:看到阳光就疼痛这个世界上有些人必须用非常极端的方法来避免阳光的照射。暴露在阳光下,会令他们的皮肤起水泡。一旦被暴露在阳光下,他们中的很多人的皮肤就会感到疼痛和灼热。这种症状像吸血鬼一样,因此人们称此病为吸血鬼症。

行尸走肉症:相信自己已经死去这种症状有高度自杀倾向和抑郁倾向,在患病期间,患者会抱怨正在失去他们的一切,包括部分或整个的身体和灵魂。他们认为自己已经死了,并且像一个死尸一样行走。这种幻觉一般扩大到一定程度,病人可以感觉到蛆虫在身体里蠕动,并且闻到尸体腐臭的味道

歇斯底里症:有奇怪的反射这种病症的原有特性表现是,病人受到任何意想不到的视觉或听觉刺激后会异常震惊,比有人鬼头鬼脑地跟在你身后还要震惊,这一症状的病害者会大声哭闹、抽打自己的身体并且絮絮叨叨。这一病症首先在缅因州的法裔加拿大伐木工人身上发现,这种个例的反射也被鉴别成全球其他地区。

Blaschko斑纹症:全身长满条纹Blaschko斑纹症是非常非常奇怪的一种症状,1910年被一位德国皮肤科学者Alred Blaschko在人体解剖中发现。Blaschko斑纹,在人类的DNA里建立起隐性的图案。很多皮肤和粘膜的先天和遗传性疾病会依照这图案表现出来。这就形成了人身体上的斑纹。
【篇二】罕见疾病
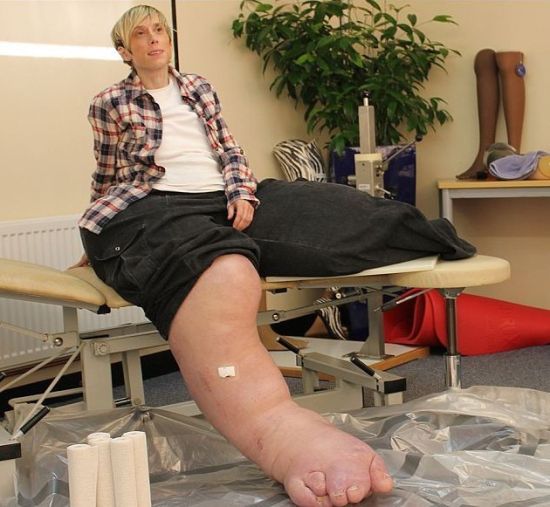
小伙患罕见疾病 右脚能穿58码的鞋子 重庆21岁的刘会长同时患有巨肢症和巨趾症,右脚整个比左脚大一倍有余,平时走路不稳,生活也多有不便,还曾被同学嘲笑是赤脚大仙。如今,完成了第一次手术的他前脚掌削短了10厘米,他还将面临两次手术。
据悉,小伙从小就发现自己的脚与常人不同,由于家庭原因也没有接受正规治疗,随着自己的年龄增长脚也不断长大。

21岁的刘会长同时患有巨肢症和巨趾症,父子俩用手比量鞋子。

病房内患病小伙的右脚用纱布包裹着,病床旁边摆放了一双大小差异巨大的鞋子,其中一只鞋子长度近40厘米。
【篇三】罕见疾病
女孩患罕见疾病 一天昏厥6次 来自英国艾塞克斯郡罗姆福的21岁年轻女孩患罕见疾病,自16岁发病以来每天突然昏厥次数高达6次,这是一种罕见血管迷走神经晕厥症(Vasovagal Syncope)。来自英国艾塞克斯郡罗姆福的21岁年轻女孩患罕见疾病,自16岁发病以来每天突然昏厥次数高达6次,这是一种罕见血管迷走神经晕厥症(Vasovagal Syncope)。

由于这种昏厥没有任何预兆,所以Jordan不能孕育孩子,因为在昏厥过程中可能会出现挤压婴儿导致婴儿窒息的现象。

在2010年圣诞节,Jordan被诊断出患有Vasovagal Syncope,字面来说就是昏厥症,所以尚不清楚昏厥的根本原因。这种突然昏厥症状使得Jordan需要一直有人陪在身旁。

Jordan很痛苦,"昏厥症阻碍了我的人生,丢了工作、不能上大学、甚至不能生孩子,因为我自己本身就是一个大隐患"。Jordan不顾一切的寻求治疗方法,想要过上正常女孩的生活,但尝试了多种药物治疗还是没有发现有效的治疗方法。

女孩患罕见疾病之后都需去医院进行X射线、CT扫描和MRI扫描,以预防器官受损,现在,Jordan在男朋友Charlie Fiander的陪伴下保持积极的心态,继续寻求治疗方法,Jordan表示在有好转之后想要研究心理学或者医学。Jordan目前正在等待一个视频遥测技术测验,即可以在昏厥过程中监控她的心脏和大脑,以寻找昏厥的根本原因,等待时间约为40周。

【篇四】罕见疾病
胆小慎看!盘点全球罕见的怪病 生命是伟大的,生命也是渺小的,健康是人生命的基本,但是现在基因遗传、病毒感染、环境危害等损害健康的因素也越来越多,世界上存在很多发病率极低,症状罕见的疾病,其中有些可以通过治疗康复,有些甚至没有有效的治疗方法。下面盘点全球罕见的怪病。1.罕见巨指症
印度小男孩穆罕默德·卡利姆(Mohammad Kaleem),从小罹患巨指症,双手因此异常肿大。
因为卡利姆异于常人的巨大双手,他经常被同龄人甚至是村民们取笑,人们称他为“被诅咒的小孩”。随着年龄的增大,卡利姆的手臂和手也随之增大,他甚至不能自己穿衣服和吃饭。

【篇五】罕见疾病
10岁女孩脸部塌陷 因患有罕见疾病 女孩脸部塌陷怎么回事?来自美国马萨诸塞州的阿利维娅?麦凯(Alivia McKay)在幼时被确诊患有帕里龙贝格综合症,化疗药物没有使病情得到改善,为此她的家人创建YouCaring帐户来筹集资金。
在2岁时,阿利维亚的父母发现她眼睛下面变得红肿,去医院检查后被诊断患有帕里龙贝格综合征。这种罕见的疾病是由皮肤下层组织的持续收缩和退化引起的,在全球范围都很罕见。
直至10岁,医生通过化疗药物控制住了病情的恶化,但表示由于青春期等生理因素必须停止使用化疗药物。现在阿利维亚只能寄希望于威斯康星州的整形外科医生约翰?西伯特博士(Dr John Siebert)的微血管游离组织转移手术。研究者表示可以用基因测序的方法,用患者身上的健康组织来代替面部的疾病组织。
由于5万美元的高昂手术费,阿利维亚的父母创建了“脸谱”和YouCaring帐户来帮助她筹集手术费用以及额外的医疗费用,到目前为止已筹集到3.4万美元。罕见疾病
【篇六】罕见疾病
全球十大罕见怪病症阅读: 189次 大小: 3KB(共2页)
疾病的存在及其分化,可能引导我们穿越整个生命过程。有超过一百万的人们在遭受以下10种罕见疾病的困扰。
1、行尸走肉症:相信自己已经死去
这种症状有高度自杀倾向和抑郁倾向,在患病期间,患者会抱怨正在失去他们的一切,包括部分或整个的身体和灵魂。他们认为自己已经死了,并且像一个死尸一样行走。这种幻觉一般扩大到一定程度,病人可以感觉到蛆虫在身体里蠕动,并且闻到尸体腐臭的味道。
2、吸血鬼症:看到阳光就疼痛
这个世界上有些人必须用非常极端的方法来避免阳光的照射。暴露在阳光下,会令他们的皮肤起水泡。一旦被暴露在阳光下,他们中的很多人的皮肤就会感到疼痛和灼热。这种症状像吸血鬼一样,因此人们称此病为吸血鬼症。
3、歇斯底里症:有奇怪的反射
这种病症的原有特性表现是,病人受到任何意想不到的视觉或听觉刺激后会异常震惊,比有人鬼头鬼脑地跟在你身后还要震惊,这一症状的病害者会大声哭闹、抽打自己的身体并且絮絮叨叨。这一病症首先在缅因州的法裔加拿大伐木工人身上发现,这种个例的反射也被鉴别成全球其他地区。
4、Blaschko斑纹症:全身长满条纹
Blaschko斑纹症是非常非常奇怪的一种症状,1910年被一位德国皮肤科学者Alred Blaschko在人体解剖中发现。Blaschko斑纹,在人类的DNA里建立起隐性的图案。很多皮肤和粘膜的先天和遗传性疾病会依照这图案表现出来。这就形成了人身体上的斑纹
5、异食癖:有吃非食物的欲望
遭受异食癖折磨的人们,会有无穷尽的欲望去吃一切不能吃的“食物”,包括纸张、泥土、胶水和腐肉。健康专家们还没有发现真正的病因,也没有找到医治的方法——尽管有报告指出,这种病症的出现可能跟病人缺乏矿物质有关。
6、爱丽丝梦游仙境症:时空和身体感觉错乱
爱丽丝梦游仙境症状(AIWS),或者叫“视微症”,是神经学上的一种高度迷惑性现象,以致影响到人类的视觉感知。患此视觉疾病的人类和动物,会把部分人以及无生命目标看得比真实的要小很多。常常这些被感知的物体,会在同一时间被看来非常得远或者非常得近。例如,一辆尺寸正常的汽车,可能被看来是一个小玩具。而一只宠物狗也可能被看作只有老鼠大小。
7、蓝肤症:蓝色的人
整个十九世纪60年代,一个庞大的“蓝人”家族居住在肯德基州山上,紧挨着克里克联盟的印第安人。他们因布鲁斯而出名。尽管他们都所有这蓝色的皮肤,但是他们很少生病而且大多数人寿命超过80岁。他们的这一体征,一代一代地遗传,大多数人拥有蓝色、靛青色
、洋李色甚至于紫色的皮肤。
8、狼人症:体毛过盛
幼儿如果患有此症会长得体型巨大,肤色暗淡,多毛的斑纹覆盖在他们的脸上。这种病症被称作狼人症,是因为患者像狼人那样拥有过盛的毛发,并且没有尖锐的牙齿和爪子。
9、象皮病:肢体的一部分变肿胀、巨大
淋巴腺的丝状病变也被称为象皮病,在这张极富戏剧性的照片中我们可以很清楚地看到这个异常粗壮、肿胀的四肢和生殖器官的病例。这一病症可能是由蚊子传播的寄生虫所导致。这一病症影响了世界上120万的人口,并且有40万人病情严重。一只患病的雌性蚊子把病毒幼虫注入人体血液当中,幼虫就会生长并且存活很多年。最后这些寄生虫聚集在周围的组织上。拥有巨大肿胀的四肢、乳房和外阴,是此症的共性表现。
10、早衰症:幼儿看上去像90岁那样苍老
早衰症是由儿童遗传密码上一个微小的错误引起的。这种病症给孩子带来非常痛苦甚至是破坏性的后果。大部分带有此病症出生的儿童将会在13岁左右死去。因为他们的身体迅速会迅速老化。他们的身体发育有着老年人的症状:心脏病、秃顶、骨质疏松以及关节炎。此病症极罕见,全世界只有48个病例。有一个家庭中,就有五个儿童遭遇这样的病症。
【篇七】罕见疾病
罕见病的定义阅读: 2140次 大小: 51KB(共6页)
http://www.chinararedisease.cn/1-6-1.html
罕见病的定义
罕见疾病简称“罕见病”,又称“孤儿病”。顾名思义,罕见病是指患病率很低、很少见的疾病。世界卫生组织(WHO)将罕见病定义为患病人数占总人口的0.65‟-1‟之间的疾病或病变,而各国(地区)对罕见病认定的标准上存在一定差异。美国的罕见病定义是患病人数少于20万人(约占总人口的0.75‟)的疾病;日本的罕见病定义是患病人数少于5万人(约占总人口的0.4‟)的疾病;澳大利亚的定义是患病人数少于2000人(约占总人口的0.1‟)的疾病;欧盟的罕见病定义是患病率低于0.5‟的疾病。各国对罕见病的不同定义,与其对罕见病药物研发的激励政策及对罕见病诊疗费用的覆盖范围有关。
随着现代医学和生物学的发展,已能识别越来越多的罕见病。然而,由于不同人种的遗传背景、生活习惯及环境的不同,人群中疾病的发生率也可能不同。所以,在一个人种中被认为是罕见病的疾病,在另外人种中可能不属于该类疾病的定义范围。同时,随着人群患病率的变化,某种疾病是否属于罕见病的范围也会发生改变。例如,艾滋病在流行初期,由于发病率极低应归属于罕见病,但随着人群患病率的增加,该病在多数国家已不属于罕见病定义的范围。
中国专家对罕见病的共识:成人患病率低于五十万分之一,新生儿中发病率低于万分之一的遗传病可定为罕见遗传病。(出自中国循证儿科杂志 2011年3月第6卷第2期)
二、罕见性遗传病
多数罕见病是慢性严重性疾病,通常会危及生命。约有80%的罕见病是由遗传缺陷引起,因此罕见病一般是指“罕见性遗传病”。国际上已发现5000多种罕见病,大约占人类疾病种类的10%。约有50%的罕见病在出生时或者儿童期即可发病,病情常进展迅速,死亡率很高,给患者家庭造成的痛苦巨大。
除了其发病率极低以外,罕见性遗传病的发生、发展、遗传规律、诊断方法等与一般的遗传病均无大的差异。但是,由于它的罕见性,一般综合医院的医生通常不容易认识、诊断出遗传性罕见病。由于常有不同系统的临床表现,根据出现各系统症状的先后顺序,罕见病患者常分别到儿科、血液科、神经科、骨科等不同的科室就诊。因此,当临床医生遇到一些不能用常见病来解释的“怪病”,应该怀疑到是否有罕见病的可能,并应及时召集相关科室的医师联合会诊,以尽早做出明确诊断。
像罕见病常被称为“孤儿病”一样,治疗罕见病的药物常被称为“孤儿药”。长期以来,绝大多数制药企业考虑到罕见病药物的市场需求量小、研发费用昂贵、在未来的销售中可能无法收回研发成本等因素,不愿进行对其开发。直到近年来,随着生物技术的发展,并在美国、欧盟等国的孤儿药相关法律的保障下,国际上才研制出少数罕见病的
治疗药物,包括美国健赞公司生产和合作的治疗戈谢氏病的伊米苷酶、治疗法布雷病的阿加糖酶β、治疗粘多糖贮积症I型的a-L-艾杜糖醛酸酶、治疗粘多糖贮积症II型的艾杜糖硫酸酯酶、治疗庞贝病的a葡萄糖苷酶等。目前,世界各国的成千上万名罕见病患者正在接受这些药物治疗,使这些罕见病患者的病情及生活质量得到了大大改善,并正在像正常人一样为社会做贡献。
尽管如此,至今仍仅有极少数的罕见性遗传病有特效的治疗方法,而多数罕见病只能依靠对症治疗。而且,由于延误诊断、获得药物困难等原因,许多患者开始治疗的时间较晚,导致病变已经不可逆转,给患者本人及其家庭造成了终身遗憾。因此,早期发现、早期诊断、早期干预,是预防和治疗罕见性遗传病、防止患者发生不可逆转的严重后遗症的重要手段。
三、罕见病的分类
与一般遗传性疾病一样,目前,罕见性遗传病主要基于2007年版的国际疾病分类法(ICD-10)、根据疾病的症状及发生机制来进行临床分类。为规范国际罕见病分类方法,世界卫生组织已成立了罕见病分类顾问小组(Topic Advisory
Group),并已开始修改对罕见疾病的分类方法。新的分类方法将主要根据疾病的病因进行分类,并将在2014年公布的ICD-11中使用。
遗传性先天性代谢缺陷(属ICD-10-E类)、神经系统发育及功能异常(属ICD-10-G类)、先天性畸形和染色体异常(属ICD-10-Q类)等是较常见的罕见性遗传疾病。其中,在婴儿期内起病的遗传性代谢病,种类繁多,涉及机体各系统组织器官。一些遗传性代谢病(包括溶酶体贮积症)已有特效治疗方法,早期诊断、早期治疗对降低病死率、减少远期神经系统后遗症具有重要意义。因此,以下对此类疾病做一重点介绍。
遗传性代谢病是遗传性生化代谢缺陷的总称,是由于基因突变引起蛋白质分子在结构和功能上发生改变,导致酶、受体、载体等的缺陷,使机体的生化反应和代谢出现异常,反应底物或者中间代谢产物在体内大量蓄积,引起一系列临床表现的一大类疾病。
遗传性代谢病种类繁多,目前已达数千种,常见有400~500种。可根据先天性缺陷所累及的生化物质再对遗传性代谢病进行分类,包括溶酶体贮积症(包括各种神经鞘脂贮积症 粘多糖贮积症 粘脂贮积症等) 过氧化体病(如肾上腺脑白质营养不良 ALD) 糖代谢病(如半乳糖血症 糖原贮积症) 氨基酸和有机酸代谢病(如苯酮尿症 高血氨症等) 核酸代谢病(如Lesch-Nyhan综合征) 脂蛋白代谢病(如棘状细胞增多症) 重金属代谢病(如肝豆状核变性 Menkes病)等 约80%的遗传性代谢病属常染色体隐性遗传,其余为X连锁遗传、常染色体显性遗传或线粒体遗传。
由于单一病种患病率较低,部分遗传代谢病(包括溶酶体贮积症)属于罕见性遗传病。
(1) 遗传性代谢病常见的症状与体征
遗传性代谢病可在婴幼儿期、儿童期、青少年期发病,其临床表现有急性危象期、缓解期和缓慢进展期。急性症状和生化异常包括急性代谢性脑病、高氨血症、代谢性酸中毒、低血糖等,随年龄不同有差异,全身各器官均可受累,以神经系统以及消化系统的表现较为突出,有些有容貌异常,毛发、皮肤色素改变。部分遗传性代谢病在婴儿早期即可有临床表现(表1)。
喂养困难、食欲差、呕吐、体重不增
嗜睡、惊厥、昏迷
呼吸困难、酸中毒、过度换气
肌张力异常
肝大
皮肤病变、毛发异常
特殊尿味、汗味
黄疸
脱水、持续呕吐、电解质异常
(2) 遗传性代谢病的诊断与治疗
多数遗传性代谢病若得不到及时诊治,常可致残,甚至危及生命,给社会和家庭带来沉重负担。近几十年来,随着生化测定和基因诊断技术的不断发展,遗传性代谢病的诊治和预防水平也在不断提高。
除病史、临床症状、体征、家族史以外,遗传性代谢病的诊断需要依赖实验室检查,如尿液三氯化铁试验、尿液二硝基苯肼(DNPH)试验和乙酸试验、尿液硝普盐试验(Brand反应)、甲苯胺蓝试验等可以对某些疾病进行初步筛查。血、尿常规分析、生化检测如血糖、血气分析、肝功能、胆红素、血氨、乳酸、酮体、丙酮
酸、肌酐、尿素、电解质、钙、磷测定,有助于对遗传性代谢病作出初步的诊断或者缩小诊断范围。
遗传代谢病的确诊主要依靠代谢物的测定和酶活性测定。可根据疾病进行氨基酸分析、铜蓝蛋白、17-羟孕酮等特异性底物或者产物的测定。串联质谱技术已成为遗传性代谢病的常规诊断工具,能对一个标本一次进行30多种氨基酸、有机酸、脂肪酸代谢性疾病的检测。气相色谱-质谱联用仪的应用对诊断有机酸尿症和某些疾病有重要意义。
基因诊断对遗传病的确定和准确分型越来越重要。对于怀疑遗传性代谢病濒临死亡的婴儿,应留取适当的标本,以便进行分析,明确病因,为遗传咨询和产前诊断提供依据。
目前根据国家“母婴保健法”的规定,新生儿疾病筛查正在全国逐步推广,除了对先天性甲状腺功能减低症、苯丙酮尿症新生儿期筛查外,有的地区开展了G6PD缺乏症、先天性肾上腺皮质增生症筛查,个别城市已经开展了串联质谱新技术的遗传性代谢病筛查,大大扩大了筛查的疾病谱。通过尽早确诊和积极治疗,可大大降低遗传性代谢病的危害性。
遗传性代谢病的总体的治疗原则是减少代谢缺陷造成的毒性物质蓄积、补充正常需要物质、酶或进行基因医疗。大多数遗传代谢病以饮食治疗为主,部分疾患可通过维生素、辅酶等进行治疗。通过对症治疗许多疾患可以得到有效控制,可以正常生活、学习和工作。
【溶酶体贮积症】
溶酶体是一种细胞器,其内部含有50多种酸性水解酶,可降解各种生物大分子,如核酸、蛋白质、脂质、粘多糖及糖原等。溶酶体贮积症是指溶酶体内的酶、激活蛋白、转运蛋白及溶酶体蛋白加工校正酶的缺乏,引起溶酶体功能缺陷,导致特定生物大分子不能正常降解而在溶酶体中贮积,使得溶酶体发生肿胀,细胞功能受到严重影响,最终导致疾病的发生。自20世纪60年代发现第一个溶酶体贮积症——庞贝病(Pompe病)以来,迄今已经发现的溶酶体贮积症约达50种,常见类型有戈谢病、法布雷病(Fabry病)、粘多糖贮积症(MPS)等,除MPS II型、Fabry病和Danon肌病属于X连锁遗传外,其余均为常染色体隐性遗传。
溶酶体贮积症最常见的症状和体征包括:(1)进行性智力、活动倒退,或先有一段正常生长发育,然后出现进行性倒退;(2)发育延迟、共济失调、惊厥、无力等神经肌肉症状;(3)面容粗陋、骨骼异常、肝脾肿大等贮积性疾病的体征;
(4)不能解释的肢体疼痛及骨痛等。患者症状呈进行性加重。
酶活性测定和基因诊断是溶酶体贮积症的主要确诊方法。北京协和医学院在国内最早建立溶酶体酶活性测定方法,可以诊断20种溶酶体贮积症,现在上海新华医院等也相继建立了酶活性检测方法。
长期以来,多数溶酶体贮积症等遗传性代谢病缺乏有效的治疗方法,只能进行对症治疗。随着生物技术的发展,国际生物制药公司已研发、上市了一些遗传代谢病的特效治疗方法——酶替代疗法。目前,酶替代疗法已可以治疗至少6种溶酶体贮积症,包括:治疗戈谢病的伊米苷酶、治疗法布雷病的阿加糖酶β、治疗粘多糖贮积症I型的a-L-艾杜糖醛酸酶、治疗粘多糖贮积症II型的艾杜糖硫酸酯酶、治疗粘多糖贮积症VI 型的Naglazyme、治疗庞贝病的a葡萄糖苷酶等。这些药物的治疗原则是特异性地补充体内由于遗传缺陷造成缺乏的代谢酶,以保持体内某种底物代谢的平衡。
酶替代疗法需终生进行,其成功与否取决于疾病阶段、疾病严重程度、酶治疗剂量、抗体产生等因素。一般来讲,开始治疗的时间越早治疗效果越好,并可避免出现脏器、骨骼病变等不可逆的损害。
目前,治疗戈谢病的药物——注射用伊米苷酶已在我国注册上市。
罕见遗传病之溶酶体贮积症
在繁忙的临床工作中,您见过这样的患者吗?曾经正常生长发育的幼儿逐渐出现面容粗陋、骨骼异常、关节活动受限、肝脾肿大、智力下降;以往没有明显异常的儿童逐渐出现贫血、血小板减少、肝脾肿大、骨骼疼痛;婴儿生后不久出现严重肌张力减低、呼吸肌无力、心脏扩大;风华正茂的青少年不明原因的出现难以忍受的四肢疼痛、皮疹、腹部不适、心脏及肾脏功能不全„„当临床上遇到患者出现这些全身多系统受累、呈进行性加重、一般治疗无效的临床症状和体征时,您是否考虑过患者可能患有罕见的遗传性疾病—溶酶体贮积症(Lysosomal storage diseases, LSDs)?
溶酶体是一种细胞器,其内部含有50多种酸性水解酶,可降解各种生物大分子,如核酸、蛋白质、脂质、粘多糖及糖原等。LSDs是指溶酶体内的酶、激活蛋白、转运蛋白及溶酶体蛋白加工校正酶的缺乏,引起溶酶体功能缺陷,导致特定生物大分子不能正常降解而在溶酶体中贮积,使得溶酶体发生肿胀,细胞功能受到严重影响,最终导致疾病的发生。迄今已经发现的LSDs约达50种。根据贮积底物的不同,可将LSDs分为粘多糖贮积症(MPSI-Ⅶ型)、粘脂质贮积症(如MLⅡ、Ⅲ型)、神经鞘脂贮积症(如戈谢病、法布雷病、尼曼匹克病、球形脑白质营养不良等)、糖原贮积症Ⅱ型(又称庞贝病)等。虽然每种单个疾病均较少见,但LSDs总的新生儿人群发病率为1∶7,000~1∶8,000。除粘多糖贮积症II型、法布雷病和Danon病属于X连锁遗传外,其余LSDs均为常染色体隐性遗传病。
世界罕见疾病 常见的罕见疾病






